
图1. NextDenovo V2.0-beta.1上线Github
测序中国2019年10月18日消息,希望组面向全球释放三代测序数据高效纠错、组装软件NextDenovo最新版本V2.0-beta.1 (https://github.com/Nextomics/NextDenovo),并免费开放用于学术和其他非商业用途。据悉,本次希望组发布的最新版本NextDenovo是专为三代测序数据开发的纠错、组装软件,不但解决了现有三代测序数据组装工具资源占用大、运行时间长、组装质量不稳定的瓶颈问题,还实现了单Contig一条染色体和超大型基因组组装的突破,为利用三代数据组装基因组扫清了组装算法的障碍。
三代测序数据组装已经成为基因组De novo的主流方案,其中Nanopore的读长可达数百kb甚至超过2Mb,在解决染色体着丝粒/端粒区域、性染色体等基因组复杂区域以及复杂基因组组装问题方面,具有更大的优势[1]。前不久,加州大学圣克鲁斯基因研究所等单位的研究人员利用Nanopore的ultra-long reads成功拼接出了首个人类X染色体基因组完成图序列[2]。
但目前的组装算法仍存在较多的瓶颈,使三代测序的优势不能完全发挥。现有三代测序数据组装软件如:Falcon[3]、Canu[4]、Miniasm[5]、Wtdbg[6]等存在以下几类问题:
胡江
针对以上问题希望组胡江团队研发出专门用于三代测序数据纠错、组装的软件——NextDenovo。其包含NextCorrect和NextGraph两个模块,依次进行测序数据的高效纠错、组装,在极大减少计算资源和运行时间的情况下,仍然能够组装出高质量基因组。基于NextDenovo,希望组已经实现了小基因组物种近完成图和>10Gb基因组物种的组装工作。
NextDenovo原始数据纠错模块可对PacBio和Nanopore的三代测序原始数据进行纠错。表1为NextDenovo与现有主流三代测序数据校正工具(Canu、Falcon和Racon[7]),对不同三代测序平台(PacBio和Nanopore)数据的纠错性能比较。NextDenovo能够在极大减少运行时间的情况下,达到甚至高于与其他软件的纠错精度。
表1. 三代测序原始数据纠错软件性能评估
注:测试数据为人20号染色体PacBio和Nanopore数据各100×,运行环境CentOS Linux release 7.4.1708 (Core),128G内存,32线程(Intel(R) Xeon(R) Gold 6151 CPU @3.00GHz),参数默认。
NextDenovo组装模块是基于String graph算法,利用纠错后的三代测序数据进行基因组高效组装。之前的评测结果https://mp.weixin.qq.com/s/8P9KeTpGc5-hIpVy4T4XxQ表明利用相同的Nanopore数据,NextDenovo在组装速度、结果连续性等指标均明显优于Canu(图2)。
图2. NextDenovo与Canu用相同Nanopore数据组装结果共线性比较
表2. NextDenovo的组装案例
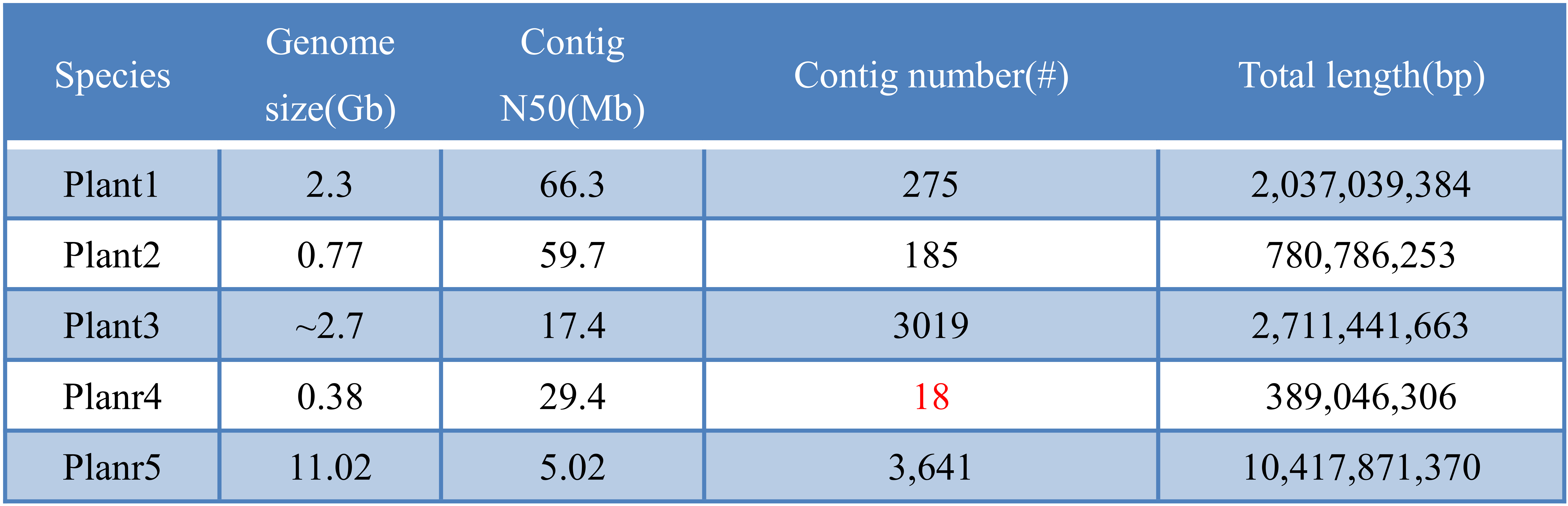
图3. NextDenovo组装基因组共线性
图4. 水稻93-11基因组不同组装结果比较
参考资料:
[1]高胜寒, 禹海英, 吴双阳, 等. 复杂基因组测序技术研究进展[J]. 遗传, 2018, 40(11): 944-963.
[2]Miga K H, Koren S, Rhie A, etal. Telomere-to-telomere assembly of a complete human X chromosome[J]. BioRxiv,2019: 735928.
[3]Chin C S, Peluso P, Sedlazeck F J, et al. Phased diploid genome assembly with single-molecule real-time sequencing[J]. Nature methods, 2016, 13(12): 1050.
[4]Koren S, Walenz B P, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation[J]. Genome research, 2017, 27(5): 722-736.
[5]Li H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences[J]. Bioinformatics, 2016, 32(14): 2103-2110.
[6]Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2[J]. BioRxiv, 2019: 530972.
[7]Sanders A D, Falconer E, Hills M, et al. Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs[J]. Nature protocols, 2017, 12(6): 1151.
[8]Zhang J, Chen L L, Xing F, et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63[J]. Proceedings of the National Academy of Sciences, 2016, 113(35): E5163-E5171.
本文由 SEQ.CN 作者:戴胜 发表,转载请注明来源!

