通过深入了解基因组产物形成的相互作用网络,人类基因组注释功能得以飞速发展。随着技术的进步,我们已经实现了DNA-DNA、蛋白质- DNA、RNA-DNA和RNA-RNA相互作用的全基因组映射图谱。但完成人类蛋白质-蛋白质相互作用(PPI)的全基因组映射图谱仍然是一项艰巨任务。
目前,大规模PPI映射方法可分为3类,即“并行化一对一”、“一对多”和“多对多”方法。“并行化一对一”方法是利用自动化和并行化来提高酵母双杂交(Y2H)分析的通量,包括高通量Y2H、MAPPIT 、QIS-seq以及将PPI对的基因序列进行融合测序的RLL-Y2H、Stitch-Seq、CrY2H-seq和BFG-Y2H等方法。“一对多”方法是从纯化或标记靶蛋白开始,在空间邻近亲和纯化、邻近生物素化(BioID)、GFP融合或蛋白微阵列中鉴定共纯化的蛋白。“多对多”方法旨在从单个实验中对所有PPI进行分析,用于解析配体-靶标对和抗体-抗原对。
上述方法也可以分为蛋白质相互作用分析和空间邻近分析。蛋白质相互作用分析可进一步分为二元和非二元分析。虽然二元分析(如Y2H)会直接产生成对蛋白质相互作用,但非二元分析(如AP-MS和co-IP)会产生物理关联,其中成对鉴定的每种蛋白质可能不会直接相互作用,例如多蛋白质复合物中的蛋白质。空间邻近性分析(例如BioID)可揭示除空间上邻近之外,可能不会形成物理相互作用或关联的蛋白质。
为高通量识别二元和多元蛋白质相互作用,加州大学圣地亚哥分校钟声教授研究团队开发了一种用于大规模检测PPI的高通量“多对多”非二元分析方法PROPER-seq,可将独特的RNA条形码附着到每种蛋白质上,并对这些条形码进行测序。结果显示,PROPER-seq能够在单个实验中扫描10000×10000个蛋白质对,并识别二元和多重蛋白质相互作用。利用PROPER-seq方法,研究团队构建了人类 PPI参考图谱,包括210518个PPI,涉及8635种蛋白质,证明该方法是一种可在转录组水平绘制PPI图谱的高效技术。近日,该研究成果在Molecular Cell上发表,文章题为“Revealing protein-protein interactions at the transcriptome scale by sequencing”。

文章发表在Molecular Cell
研究团队设想将每个PPI转换为独特的DNA序列,然后利用DNA测序的极高通量来解码这些PPI。为此,研究人员开发了一种改进的mRNA-display方法,命名为SMART-display,可将一个独特的RNA条形码附着在每个蛋白质上。通过INLISE方法,对附着在两个相互作用的蛋白质上的一对DNA条形码进行测序。研究团队将结合SMART-display和INLISE的整体方法命名为PROPER-seq。
图1. 结合SMART-display和INLISE的整合图。来源: Molecular Cell
SMART-display的输入是用户选择的一组细胞。SMART-display的中间产物是一个适配于mRNA-display的基因库,其中转录起始、翻译起始和嘌呤霉素连接的序列被整合到每个基因的适当位置(图2)。SMART-display输出的是一个mRNA-连接蛋白复合体形式的display库。
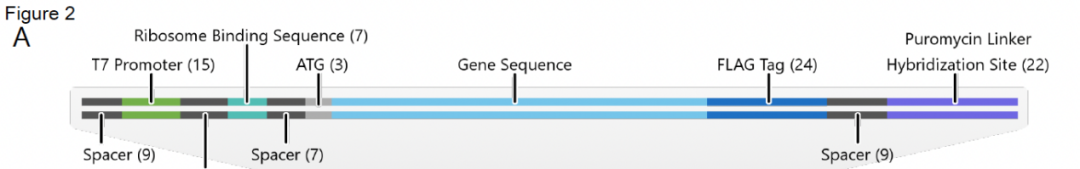

图2. 转录起始、翻译起始和嘌呤霉素连接的序列位置图。来源: Molecular Cell
INLISE是PROPER-seq关键的第二步,将PPIs转换成结构为cdna1 - linking - cdna2的嵌合序列(图3)。INLISE的输入是SMART-display生成的两个display库。每个库包含大约30000个mRNA-蛋白复合体。其中一个文库称为“诱饵”文库,通过嘌呤霉素连接序列上的生物素被固定在链霉亲和素珠上。另一个库称为“猎物”库,与诱饵库混合可发生相互作用。去除假的相互作用后,相互作用蛋白的mRNA条形码被连接,以cDNA1- linking -cDNA2的形式创建一个嵌合序列,其中cDNA1和cDNA2代表两个相互作用的蛋白。这些嵌合序列随后被选择并进行对端测序。
图3. cdna1 - linking - cdna2的嵌合序列图。来源: Molecular Cell
为了绘制人类PPI网络,研究团队将PROPER-seq应用于人胚胎肾细胞、T淋巴细胞和内皮细胞,将构建的6个PROPER-seq库(HEK1、HEK2、JKT1、JKT2、HUVEC1和HUVEC2)合并为一个数据集。该数据集由大约14亿对reads组成,包括210518个成对的PPIs,涉及8635个蛋白质,统称为PROPER v1.0网络(图4A)。其中,1,365 和2,480个PPI得到已发表的co-IP和AP-MS数据的支持,17,638个由PrePPI算法预测但未经此前实验验证,并与100个与人类合成致死基因对重叠。分析数据显示,PROPER v1.0的测序reads覆盖16305个人类蛋白编码基因,其中8635个蛋白编码基因参与了PROPER v1.0的PPIs。通过比较3对二元和非二元PPI,发现非二元PPIs在PROPER v1.0富集,表明PROPER v1.0同时包含二元和非二元PPIs。此外,研究团队还使用接近连接试验(PLA)和co-IP对未被描述过的4个PPIs进行了实验验证。
图4. PROPER v1.0网络图。来源: Molecular Cell
在评估PROPER v1.0后,研究团队测试了细胞类型特异性基因表达是否会导致每种细胞类型的PROPER-seq数据对PROPER v1.0中鉴定的PPIs的贡献存在差异。研究人员在两个层次上测试了这种可能性,即每个PPI和每个子网的GO富集。在单个PPIs的水平上,约33%的PROPER v1.0的PPIs主要是由于来自特定细胞类型的reads对被识别出来的 (图6A)。在子网层面,大多数子网络并不优先与三种特定细胞类型中的任何一种相关联(图6B)。以上数据表明,PROPER-seq分析对于揭示细胞类型特异性PPIs具有很强的潜力。
图5. 子网络关联图。来源: Molecular Cell
综上,在单个实验中,PROPER-seq是一种可在转录组水平绘制PPI网络的高效快速的方法,可以应用于各种细胞,且不需要专门的资源或试剂,如抗体。同时,PROPER v1.0 数据库提供了约200000个未表征的PPI,扩展了人类参考蛋白质相互作用数据。值得注意的是,PROPER v1.0为超过17000个未经实验验证的计算预测的PPI提供了实验支持,表明基于结构的计算模型具有很强的预测能力。研究团队表示,利用PROPER-seq,普通实验室在几周时间内就能获得目的细胞的整个蛋白相互作用网络,帮助发现与许多目的细胞或组织相关的PPI。
本文由 SEQ.CN 作者:白云 发表,转载请注明来源!

